
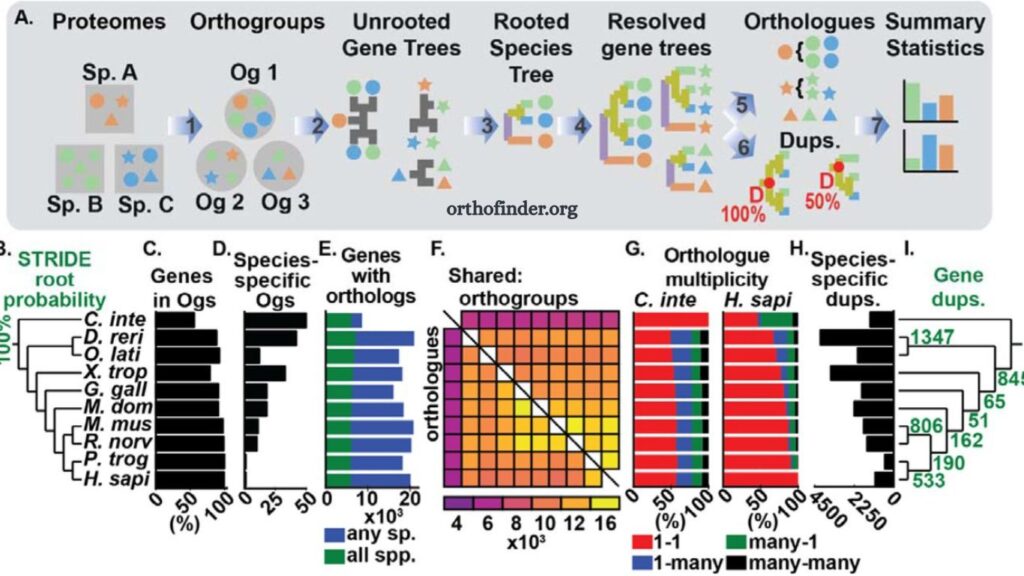
Comparative genomics depends on the accurate identification of orthologous genes across different species. OrthoFinder has become one of the most reliable tools for this purpose due to its speed, accuracy, and ability to handle large-scale datasets. Researchers use it to analyze evolutionary relationships, gene family expansions, and functional genomics across multiple species.
Running OrthoFinder on multiple species datasets requires proper data preparation, correct execution steps, and an understanding of its output structure. This guide explains the complete workflow in a clear, practical, and SEO-friendly way.
What is OrthoFinder?
OrthoFinder is a bioinformatics software tool designed to identify orthogroups—sets of genes descended from a single gene in the last common ancestor of the species being studied. It also constructs gene trees and infers species trees, making it highly valuable for evolutionary and comparative genomic studies.
Unlike basic similarity tools, OrthoFinder uses advanced algorithms to reduce bias and improve orthology inference across multiple species simultaneously.
Read More: OrthoFinder Analysis: Input Files You Need to Get Started
Why Use OrthoFinder for Multiple Species Analysis?
Multi-species genomic analysis requires tools that scale efficiently and maintain accuracy. OrthoFinder provides several advantages:
- High accuracy in ortholog detection
- Scalable performance for large datasets
- Automated gene and species tree inference
- Minimal manual configuration
- Compatibility with protein sequence datasets
These features make it ideal for studies involving evolutionary biology, plant genomics, animal genetics, and microbial comparisons.
Preparing Data for OrthoFinder
Proper input preparation determines the success of OrthoFinder analysis. Each species must be represented with a separate protein FASTA file.
Step 1: Collect Protein Sequences
Download protein FASTA files for all species under investigation. Databases such as Ensembl, NCBI, or UniProt provide reliable datasets.
Step 2: Organize Directory Structure
Create a dedicated folder for the analysis. Place each species’ FASTA file inside this directory.
Example structure:
- project_folder/
- ├── species1.faa
- ├── species2.faa
- ├── species3.faa
Each file must contain only protein sequences. Mixing nucleotide sequences will lead to incorrect results.
Step 3: Validate File Format
Ensure FASTA headers are unique and properly formatted. Remove duplicate identifiers and invalid characters. Clean data improves clustering accuracy during analysis.
Installing OrthoFinder
OrthoFinder runs on Linux, macOS, or Windows (via WSL or Conda environments).
Using Conda (Recommended Method)
Install OrthoFinder using the following command:
conda install -c bioconda orthofinder
This method automatically handles dependencies such as DIAMOND and MCL.
Manual Installation
Download OrthoFinder from its official repository and ensure dependencies are installed separately. Manual setup requires more configuration but offers flexibility.
Running OrthoFinder on Multiple Species Datasets
After preparing data and installing the software, the main execution step is straightforward.
Basic Command
Navigate to the folder containing FASTA files and run:
orthofinder -f /path/to/project_folder
OrthoFinder automatically performs the following steps:
- Sequence similarity search
- Orthogroup inference
- Gene tree construction
- Species tree inference
- Functional annotation mapping (optional)
Understanding Output Files
OrthoFinder generates multiple output directories. Each plays a role in interpreting evolutionary relationships.
Orthogroups Folder
Contains gene clusters shared across species. These represent sets of orthologous and paralogous genes.
Gene Trees Folder
Includes phylogenetic trees for each orthogroup. These trees help study gene evolution.
Species Tree Folder
Represents evolutionary relationships between the studied species.
Working Directory
Stores intermediate files used during computation. These help in debugging and re-analysis if needed.
Running OrthoFinder with Multiple Threads
Large datasets benefit from parallel processing. OrthoFinder supports multi-threading to reduce runtime.
Command with Threads
orthofinder -f /path/to/project_folder -t 16
Higher thread counts improve performance but require sufficient CPU resources.
Using Advanced Options in OrthoFinder
OrthoFinder provides several advanced parameters for customized analysis.
Using DIAMOND for Faster Searches
orthofinder -f /path/to/project_folder -S diamond
DIAMOND significantly speeds up similarity searches without compromising accuracy.
Memory Optimization
For large datasets:
orthofinder -f /path/to/project_folder -op
This option optimizes memory usage during computation.
Resume Analysis
Interrupted runs can be resumed:
orthofinder -f /path/to/project_folder -b previous_results/
Best Practices for Multi-Species OrthoFinder Analysis
Proper planning improves both speed and accuracy.
Maintain Clean Data
Remove low-quality sequences and redundant isoforms before analysis.
Limit Number of Species per Run
Very large datasets may require splitting into smaller batches for efficiency.
Use Consistent Naming
Species file names should be simple and consistent to make results easier to interpret.
Check Input Quality
Ensure each FASTA file contains only protein sequences from a single species.
Common Errors and Fixes
Error: Missing Dependencies
Install many issing tools using Conda or syour system’spackage mmanager
Error: Low Memory Issues
Reduce thread count or split the dataset into smaller groups.
Error: Invalid FASTA Format
Validate sequence headers and remove unsupported characters.
Applications of OrthoFinder in Multi-Species Studies
OrthoFinder supports a wide range of biological research applications.
Evolutionary Biology
Researchers study gene evolution and patterns of divergence across species.
Plant Genomics
Helps identify gene families responsible for traits like drought resistance and yield.
Animal Genetics
Used to compare genomes across vertebrates and invertebrates.
Microbial Research
Supports comparative analysis of bacterial and fungal species.
Performance Tips for Large Datasets
Large-scale genomic studies require optimization strategies.
- Use SSD storage for faster I/O performance
- Increase RAM allocation for large datasets
- Enable DIAMOND for faster similarity searches
- Avoid running unnecessary background processes
- Split extremely large datasets into logical groups
Frequently Asked Questions
What is OrthoFinder used for?
OrthoFinder identifies orthologous genes across multiple species and helps study gene evolution, function, and phylogenetic relationships.
Can OrthoFinder handle multiple species at once?
Yes, OrthoFinder is specifically designed to efficiently analyze multiple-species datasets in a single run.
What input files does OrthoFinder require?
OrthoFinder requires protein FASTA files, with one file per species containing all protein sequences.
How long does OrthoFinder take to run?
Runtime depends on the dataset size, the number of species, and the available computing power, ranging from minutes to several hours.
Can OrthoFinder run on Windows?
Yes, but it works best on Linux or macOS. On Windows, it can be used through WSL or Conda environments.
What are orthogroups in OrthoFinder?
Orthogroups are sets of genes from different species that evolved from a single ancestral gene.
How can I speed up OrthoFinder analysis?
Using multi-threading, DIAMOND for sequence searches, and high-performance hardware significantly improves runtime.
Conclusion
OrthoFinder delivers a robust and efficient solution for multi-species genomic analysis. Proper dataset preparation, correct installation, and optimized execution ensure accurate ortholog detection and reliable evolutionary insights. Its ability to process multiple species simultaneously makes it highly valuable for comparative genomics, evolutionary biology, and functional gene studies. Consistent workflow practices and appropriate parameter selection further enhance performance and result quality.