
Modern comparative genomics relies heavily on accurate identification of gene relationships across different species. Researchers often need to understand which genes share a common ancestry and how they evolved. OrthoFinder has become one of the most widely used tools for solving this challenge with high accuracy and efficiency. Designed for large-scale genomic analysis, it helps scientists detect orthologs, infer gene trees, and reconstruct evolutionary relationships across multiple species.
Growing genomic datasets demand tools that combine speed, precision, and scalability. OrthoFinder addresses these needs by automating complex steps in orthology inference, making it a key resource in bioinformatics and evolutionary biology.
Understanding OrthoFinder
OrthoFinder is a bioinformatics software package that identifies orthologous genes across multiple species. Orthologs are genes in different organisms that originated from a common ancestral gene through speciation events. These genes usually retain similar functions, making them essential for evolutionary studies and functional annotation.
Unlike traditional approaches that rely solely on pairwise comparisons, OrthoFinder uses a comprehensive method that integrates sequence similarity, phylogenetic tree construction, and clustering algorithms. This allows it to produce more accurate and biologically meaningful results.
Read More: What is OrthoFinder and how does it work?
Why OrthoFinder Matters in Genomics
Comparative genomics depends on accurately identifying gene relationships. Incorrect classification of genes can lead to misleading conclusions in evolutionary studies, disease research, and functional genomics.
OrthoFinder improves research outcomes in several ways:
- Enhances accuracy in ortholog detection
- Reduces computational bias seen in older methods
- Handles large genomic datasets efficiently
- Supports multi-species comparisons at scale
Researchers working on plant genomics, animal evolution, and microbial diversity frequently rely on OrthoFinder to generate reliable gene family data.
Core Concept Behind OrthoFinder
Gene evolution occurs through two major processes: speciation and duplication. Speciation separates genes into different species, while duplication creates gene copies within the same genome.
OrthoFinder distinguishes between:
- Orthologs: Genes separated by speciation
- Paralogs: Genes created by duplication events
Understanding this distinction is critical because orthologs often retain similar biological functions, while paralogs may evolve new roles.
OrthoFinder uses evolutionary principles to reconstruct gene histories and classify relationships correctly.
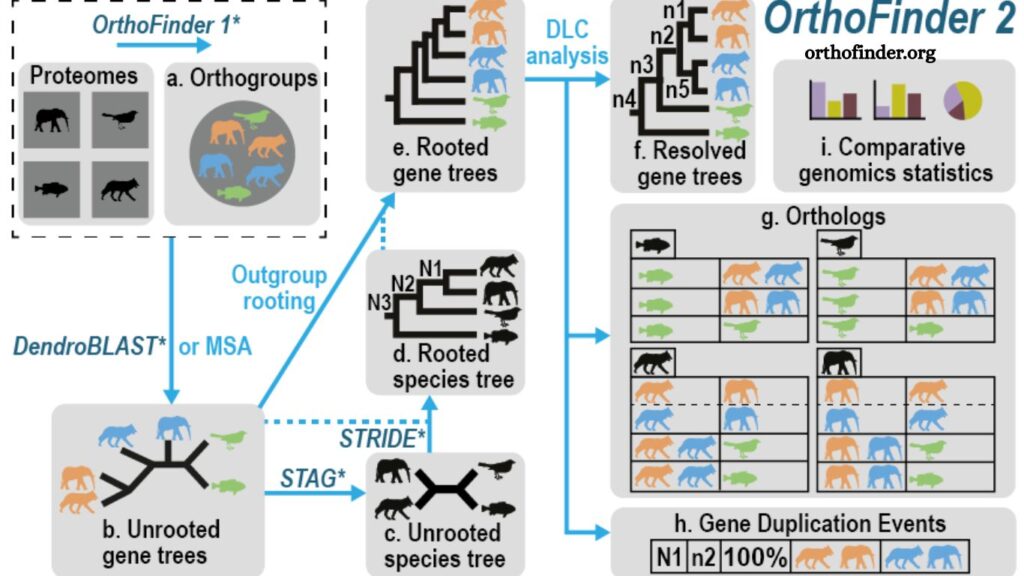
How OrthoFinder Works
The OrthoFinder workflow follows a structured pipeline that transforms raw protein sequences into evolutionary insights.
Input Preparation
Researchers provide protein sequences from multiple species. These sequences typically come from genome annotation files in FASTA format. OrthoFinder processes these sequences as the starting point of analysis.
Sequence Similarity Search
The software performs all-vs-all sequence comparisons using fast alignment tools. This step identifies similarities between genes across different species.
Sequence similarity underpins the grouping of genes into potential families.
Orthogroup Construction
Genes are clustered into orthogroups based on similarity patterns. An orthogroup contains genes that originated from a single ancestral gene in the last common ancestor of all included species.
This step ensures that related genes are grouped before deeper evolutionary analysis begins.
Gene Tree Inference
Each orthogroup undergoes phylogenetic tree construction. These gene trees represent evolutionary relationships between genes, showing how they diverged over time.
Tree-building methods in OrthoFinder are optimized for both accuracy and computational efficiency.
Species Tree Estimation
Using information from gene trees, OrthoFinder reconstructs a species tree. This represents the evolutionary relationships among the studied species.
Unlike traditional methods that require predefined species trees, OrthoFinder can infer them automatically.
Ortholog Identification
Final orthologs are identified by reconciling gene trees with the species tree. This process distinguishes speciation events from duplication events, ensuring precise classification.
The result is a comprehensive map of orthologous relationships across species.
Key Features of OrthoFinder
Several features make OrthoFinder a preferred tool in genomic research:
- High accuracy in orthology inference
- Automated species tree reconstruction
- Scalable architecture for large datasets
- Integrated gene tree analysis
- Minimal parameter tuning required
- Compatibility with multiple genome formats
These features reduce manual effort and improve reproducibility in research workflows.
Applications of OrthoFinder
OrthoFinder supports a wide range of biological and computational studies:
Comparative Genomics
Researchers use it to compare genomes across species and identify conserved genes.
Evolutionary Biology
The tool helps trace gene evolution and understand species divergence.
Functional Genomics
Ortholog detection helps predict gene functions in newly sequenced organisms.
Agriculture and Crop Research
Scientists analyze plant genomes to improve crop traits and disease resistance.
Medical Genomics
Understanding gene evolution supports research into genetic disorders and drug targets.
Advantages of Using OrthoFinder
OrthoFinder offers several advantages over traditional orthology tools:
- Faster processing for large datasets
- Reduced false positives in gene clustering
- Improved evolutionary accuracy
- Automated pipeline from input to final results
- Strong community adoption and validation
These benefits make it suitable for both small-scale studies and large genome projects.
Limitations of OrthoFinder
Despite its strengths, OrthoFinder has certain limitations:
- Requires high-quality genome annotations
- Performance depends on input sequence accuracy
- Computational demand increases with extremely large datasets
- Interpretation of results may require biological expertise
Understanding these limitations helps researchers use the tool more effectively.
Practical Workflow Example
A typical OrthoFinder analysis follows this sequence:
- Collect protein sequences from multiple species
- Run similarity searches to identify gene relationships
- Cluster genes into orthogroups
- Build gene trees for each group
- Infer species tree from gene data
- Extract ortholog relationships for downstream analysis
This automated workflow saves significant time compared to manual methods.
Role of OrthoFinder in Modern Bioinformatics
Advancements in sequencing technologies have led to an explosion of genomic data. Manual analysis methods cannot handle this scale efficiently.
OrthoFinder plays a critical role in modern bioinformatics by providing an automated and statistically robust framework for orthology inference. Researchers rely on it to generate insights that drive discoveries in evolution, genetics, and molecular biology.
Frequently Asked Questions
What is OrthoFinder used for?
OrthoFinder is used to identify orthologous genes across multiple species and analyze their evolutionary relationships in genomic studies.
How does OrthoFinder define orthologs?
It defines orthologs as genes that originate from a common ancestor through speciation events and often retain similar biological functions.
Is OrthoFinder suitable for large datasets?
Yes, OrthoFinder is designed to handle large genomic datasets efficiently with high accuracy and scalable performance.
Does OrthoFinder require a species tree beforehand?
No, OrthoFinder can automatically infer a species tree using gene trees generated during the analysis.
What input does OrthoFinder need?
It requires protein sequence files, usually in FASTA format, from different species for comparative analysis.
What makes OrthoFinder different from other tools?
OrthoFinder combines sequence similarity, gene tree construction, and species tree inference for more accurate orthology prediction.
Can OrthoFinder be used in medical research?
Yes, it supports medical genomics by helping identify gene functions and evolutionary links relevant to diseases and drug research.
Conclusion
OrthoFinder provides a powerful and reliable approach for identifying orthologous genes and reconstructing evolutionary relationships across multiple species. Its automated workflow, high accuracy, and ability to handle large genomic datasets make it a valuable tool in modern bioinformatics. Researchers use it to simplify complex comparative genomics tasks and gain clearer insights into gene evolution, functional annotation, and species divergence.